1. Historical
The hydrogenation process of edible oils was invented by Wilhelm Normann in 1902 [1]. At that time, it was possible to follow the progress of the reaction by measuring the iodine value of the reaction product. Measuring its melting point also provided a way of characterisation and measuring both soon revealed that when a certain oil was partially hydrogenated to a certain melting point, its iodine value could vary. This led to the concept of selectivity, be it as yet poorly defined.
In the mid-1900s, determining a fatty acid composition was laborious, not very accurate and far less detailed than current analyses based on capillary GLC. Even so, an attempt to facilitate the understanding of what happens during a hydrogenation reaction was based on the fatty acid composition of the hydrogenation product [2]. It proposed the working hypothesis that the rate of reaction of a fatty acid in a triglyceride molecule did not depend on the chemical nature of the other fatty acids in said molecule. This so-called “common fatty acid pool” concept has been most useful for half a century when it was demonstrated to be incorrect [3].
To describe what happens during a hydrogenation run, a number of different selectivities have been defined:
- Positional selectivity. It is not self evident that an unsaturated fatty acid esterified to the central hydroxyl group of the glycerol backbone will react at the same rate as those attached to the outer hydroxyl groups. Some early authors noted small differences but nowadays, there is a consensus that the positional selectivity does not exist [3].
- Linole(n)ic acid selectivity. This selectivity indicates to what extent the reaction of the polyunsaturated linoleic acid prevails over the reaction of the monounsaturated oleic acid. It has therefore been defined as a ratio of rate constants. Since both reactions were assumed to depend in identical manner on the hydrogen concentration, the linoleic acid selectivity was defined as a ratio of the hydrogenation rate constants of linoleic acid and oleic acid and determined as the ratio of the relative rates of reaction of linoleic acid and oleic acid, whereby the relative rate was defined as the rate of disappearance of a reagent divided by its concentration. Similarly, the linolenic acid selectivity was defined as the ratio of the hydrogenation rate constants of linolenic acid and linoleic acid.These selectivities have been found to be very useful in describing and comparing hydrogenation processes and catalysts. However, the assumption underlying their definition that both reactions depend in the same way on the hydrogen concentration turned out to be wrong. This was demonstrated by the fact that the selectivity, defined as a ratio of two constants, is not constant during a hydrogenation run [3].
- trans Selectivity. This selectivity has been defined as the ratio of the content of trans fatty acids and the decrease in iodine value of an oil being hydrogenated. When these variables are plotted against each other, a line results that is straight for a surprisingly long part of the hydrogenation run. Only when oleic acid and elaidic acid are beginning to be hydrogenated does this line bend downwards to reach zero in the fully hydrogenated product when the iodine value has reached zero as well.
- Triglyceride selectivity. This concept was introduced by Coenen [4] to explain why some wide-pore hydrogenation catalysts can yield different hydrogenation products from narrow-pore catalysts. A way to quantify this selectivity has been proposed but it has hardly been used.
Other ways to describe what happens during a hydrogenation have comprised the introduction of the concept of shunt reactions. Accordingly, the oleate shunt assumed that a linolenic acid moiety reacted to form oleic/elaidic acid without forming a di-unsaturated intermediate [5]. Rate constants were estimated from analogue (!) computer simulations but forgotten about when more reliable fatty acid compositions became available. Another conclusion that turned out to be untenable was that monoenoic acids are always being saturated when they are being isomerised [6]. It was based on the observation that both behenic acid (C22:0) and trans-brassidic acid (trans-C22:1) were formed when high erucic acid rapeseed oil was partially hydrogenated. However, when the reaction was repeated under selective conditions at much higher temperature, trans-brassidic acid was formed without behenic acid being formed (W.L.J. Meeussen, personal communication).
2. Currently Accepted Hydrogenation Mechanism
It is now commonly accepted that the nickel-catalysed hydrogenation of unsaturated fatty acids follows the Horiuti-Polanyi mechanism. According to this mechanism, molecular hydrogen is adsorbed onto the nickel surface (reaction 1 in the figure below where adsorbed species are indicated by an asterisk) and dissociated into two hydrogen atoms (reaction 2). Fatty acids are also adsorbed onto this nickel surface by their double bond or bonds and in a first step, a hydrogen atom is added to this bond to form a half-hydrogenated intermediate. If a second hydrogen atom is then added to this intermediate, the original double bond has been saturated but because the first addition is reversible, the intermediate can also dissociate.
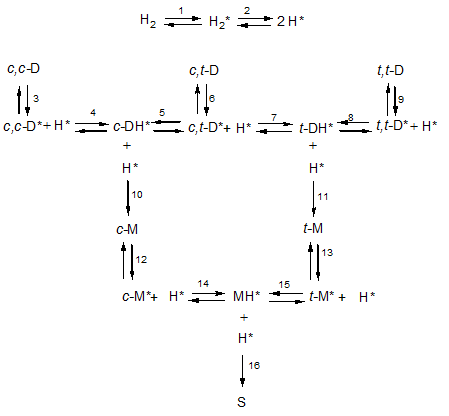
Such a reaction scheme is shown in the figure above. It shows how dienes (D for short) are hydrogenated to from monoenes (M) and finally stearic acid (D). So linoleic acid (9c,12c-octadecadienoic acid, c,c-D) is reversibly adsorbed in reaction 3 and a hydrogen atom H* is reversibly added to the adsorbed linoleic acid (c,c-D*) to form a half-hydrogenated intermediate (c-DH*). This is still adsorbed as shown by the asterisk (*) but has only a single double bond left that has retained its cis-configuration. This half-hydrogenated intermediate can do one of several things. It can react irreversibly with a further hydrogen atom (H*) in reaction 10 or it can dissociate.
The hydrogen atom leaving on this dissociation can be the same as the one that has been added as shown in reaction -4 (where the minus sign indicates the reverse reaction), it can be a different atom on the same carbon atom or it can be a hydrogen atom leaving from a different carbon atom. Accordingly, the fatty acid resulting from this dissociation can have undergone geometrical isomerisation so that the original cis-configuration of the double bond has been changed into a trans-configuration as shown in reaction 5. It can also have undergone positional isomerisation meaning that the double bond has shifted one position along the fatty acid chain; this type of isomerisation is not shown in the figure. In methylene-interrupted polyunsaturated fatty acids this can lead to the formation of conjugated double bonds. Because the double bond is no longer present in the half-hydrogenated intermediate, this intermediate can rotate around the original double bond and this can result in an isomerisation that is both geometrical and positional.
When a half-hydrogenated intermediate is saturated by reacting with another hydrogen atom, heat is liberated since the hydrogenation process is strongly exothermic. Consequently, the reaction product is so ‘hot’ that it immediately leaves the catalyst surface. By sharing its kinetic energy with its surroundings, it cools down so that it can be re-adsorbed at its remaining double bond (c-M*). This monoene can then react with an adsorbed hydrogen to form the half-hydrogenated monoene (MH*) that just as above can react in several ways one of which (reaction 16) leads to stearic acid.
3. Kinetics of the Hydrogenation Process
The mechanism outlined above shows which reactions can take place. The kinetics of the process determine which ones actually take place. Consequently, the kinetics of the hydrogenation process govern the hydrogenation product properties and controlling these properties amounts to changing the kinetics of the process.
The adsorption (reaction 1) and dissociation (reaction 2) of the hydrogen are governed by two factors: the catalyst (type and amount) and the concentration of the hydrogen in the bulk of the oil. Noble metal catalysts differ from the commonly used nickel catalyst, and amongst nickel catalysts, the manufacturers produce various types. One of these types is a trans-promoting catalyst. It has been poisoned with sulphur with the result that the surface concentration of active sites has been decreased which in turn also causes a decrease in the surface concentration of the hydrogen atoms (H*). Consequently, the dissociation reactions 14 and 15 of the monoene (MH*) are favoured kinetically over its saturation (reaction 16) since the latter requires an adsorbed hydrogen atom. So decreasing the surface concentration of adsorbed hydrogen atoms slows down the overall rate of reaction and causes isomerisation of double bonds.
The other factor is the concentration of the hydrogen in the bulk of the oil. This concentration is the result of a dynamic equilibrium between the hydrogen supply and its demand. The supply is proportional to the difference between the hydrogen solubility as determined by the oil temperature and the system pressure and its actual concentration. It is also a function of the agitation (type and speed of agitator) that causes the hydrogen to dissolve. The demand is proportional to the rate of hydrogenation which is governed by the amount and type of catalyst, the reactivity of the oil and the hydrogen concentration.
Temporarily stopping the agitator will cause the hydrogen still present to react so that its concentration will drop and the rate of hydrogenation will slow down. Switching it on again will cause hydrogen to dissolve at a somewhat faster rate than before it was switched off since the difference between the hydrogen solubility and its concentration has increased. A lower rate of reaction and a higher rate of hydrogen dissolution will cause the hydrogen concentration to increase. That increase causes the rate of reaction to increase but the rate of dissolution to decrease until both rates are equal again. During a hydrogenation run, equipment and process parameters like the agitator, the amount of catalyst or the pressure do not change. This means that during such a run the hydrogen concentration is determined by the reactivity of the system as reflected by its fatty acid composition.
The reaction of the unsaturated fatty acids starts with their being adsorbed onto the catalyst surface. We know that polyunsaturated fatty acid are adsorbed since they are saturated and that monounsaturated ones are also adsorbed since they isomerize. Their being part of a triacylglycerol molecule means that this molecule is adsorbed onto this surface. The number of the unsaturated fatty acids in this molecule and their degrees of unsaturation determine the likelihood of this adsorption that can be quantified in a triglyceride-based system as the frequency factor of the rate constant of adsorption of a particular triglyceride.
As proposed earlier [7], the frequency factor pertaining to a monounsaturated triglyceride such as P2O or S2O (P = palmitic acid; O = oleic acid; S = stearic acid) could be arbitrarily set at o. The triglycerides like PO2 and SO2 might show a frequency factor of their rate of adsorption equal to o x s, where 1 < s < 2. For trioleate the frequency factor might well be equal to o x s2. But what about triglycerides containing an elaidic moiety?
The cis-trans equilibrium is governed by an enthalpy difference of ΔH = 4 kJ/mol. This means for instance that at 100°C, the equilibrium corresponds to 79% trans, whereas at 250°C the value is only 72%. In the figure, the half-hydrogenated intermediate MH* has an equal likelihood of reacting according to reaction –14 or reaction 15. Consequently, the position of this equilibrium must originate from a preceding reaction. Since it is enthalpy related, it is likely to originate from a difference in activation energy. Accordingly, the frequency factor of the elaidate moiety is likely have the same value as the oleic acid one and equal o as well.
To triglycerides containing a single linoleic acid moiety like P2L and S2L (L = linoleic acid) a frequency factor l can be assigned. Since linoleic acid has two double bonds and oleic acid only one, it is likely that l > o but it is not known how much larger. In fact, l and o could be almost equal since the large difference in behaviour between linoleic acid and oleic acid (the so-called linoleic acid selectivity) is likely to stem from the difference in behaviour between their half-hydrogenated intermediates (c-DH* and t-DH* versus MH*), which have one versus no double bond left.
Once the diene has been adsorbed onto the catalyst surface, it reacts with a hydrogen atom and to what extent it dissociates or reacts with a further hydrogen atom is affected by the concentration [H*] of the hydrogen atoms on the catalyst surface, whereby a high concentration favours saturation. This holds for the dienes (c-DH* and t-DH*) but also for the monoene (MH*). However, the hydrogen concentration is not the only factor affecting this extent. It is also affected by the rates of dissociation and these are governed by the respective rate constants whereby k-4 = k5 << k-14 = k15.
Accordingly, half-hydrogenated dienes remain on the catalyst surface and just wait there until they are further hydrogenated. This explains why partially hydrogenated vegetable oils hardly contain dienes with trans double bonds (c,t-D, t,c-D and t,t-D). Half-hydrogenated monoenes on the other hand readily dissociate again and this explains the presence of large amounts of trans monoenes. This amount can be decreased by favouring the saturation of MH* over its saturation by increasing [H*] by increasing the rate of agitation, increasing the system pressure and/or lowering the temperature of reaction.
The above kinetics also explain why the linoleic acid selectivity decreases during a hydrogenation run. This selectivity has been defined as the ratio of the rates of two saturation reactions. For the linoleic acid saturation, the rate-determining step is the formation of the half-hydrogenated intermediate. Once this has been formed, it will hardly dissociate and just wait until it reacts with a further hydrogen atom. So this latter rate is almost equal to the rate of formation of the half-hydrogenated intermediate and therefore proportional to [H*]. For the oleic acid, the situation differs in that the half-hydrogenated intermediate dissociates causing its steady state concentration [MH*] to be proportional to [H*]. The rate of the bimolecular saturation reaction can be assumed to be proportional to the concentration of the reagents, thus to [MH*] x [H*] and therefore to [H*]2. During a hydrogenation run, the reactivity of the reaction mixture decreases. Therefore, the hydrogen concentration increases causing [H*] to increase and thereby favouring reactions that depend more strongly on this hydrogen atom concentration. Accordingly, the saturation of monoenes that is suppressed by a low hydrogen concentration in the start of the reaction gets more and more favoured as and when the reaction proceeds causing the linoleic acid selectivity to decrease.
4. Discussion
What happens during a hydrogenation reaction has intrigued scientists for over a century. During this period, various hypotheses have been put forward some of which were refuted quite soon, some much later and some even survived up till now. So although insight in what happens has steadily improved, it is still far from perfect. The frequency factors l and o introduced recently have not yet been quantified and a reliable instrument to measure the hydrogen concentration in the bulk of the oil has not been developed, although its principle of operation has been disclosed.
In fact, the current trans scare may well be responsible for my impression that research on the hydrogenation process is decreasing. The number of publications on the subject certainly is. I regard this as a pity. I found working in the field of hydrogenation a highly rewarding challenge.
References
- Normann, W. (Herforder Maschinenfett-und Ölfabrik Leprince und Siveke), Process for converting unsaturated fatty acids or their glycerides into saturated compounds, British Patent 1 515 (1903).
- Bailey, A.E. and Fisher, G.S. Modification of vegetable oils. V. Relative reactivities toward hydrogenation of the mono- di- and triethenoid acids in certain oils. Oil & Soap, 23, 14-18 (1946).
- Dijkstra, A.J. Hydrogenation revisited. Inform, 8, 1150-1158 (1997).
- Coenen, J.W.E. The rate of change in the perspective of time. Chem. Ind., 709-722 (1978).
- Bailey, A.E. Some additional notes on the kinetics and theory of fatty oil hydrogenation. J. Am. Oil Chem. Soc., 26, 644-648 (1949).
- Coenen, J.W.E. and Boerma, H. Absorption der Reaktionspartner am Katalysator bei der Fetthydrierung. Fette Seifen Anstrichm., 70, 8-14 (1968).
- Dijkstra, A.J. Selectivities in partial hydrogenation. J. Am. Oil Chem. Soc., 87, 115-117 (2010) (DOI: 10.1007/s11746-009-1507-z).